
 Current research demonstrated that
machine learning can be adopted to build up high-accurate predictive
models in drugs/cyclodextrins (CDs) systems. Molecular descriptors
of compounds and experimental conditions were employed as inputs,
while complexation free energy as outputs. Results showed that the
light gradient boosting machine provided significantly improved
predictive performance over random forest and deep learning.
The mean absolute error was 1.38 kJ/mol and squared correlation
coefficient was 0.86. In conclusion, the developed predictive
models were able to quickly and accu-rately predict the
solubilizing capacity of CD systems.
Current research demonstrated that
machine learning can be adopted to build up high-accurate predictive
models in drugs/cyclodextrins (CDs) systems. Molecular descriptors
of compounds and experimental conditions were employed as inputs,
while complexation free energy as outputs. Results showed that the
light gradient boosting machine provided significantly improved
predictive performance over random forest and deep learning.
The mean absolute error was 1.38 kJ/mol and squared correlation
coefficient was 0.86. In conclusion, the developed predictive
models were able to quickly and accu-rately predict the
solubilizing capacity of CD systems.
Properties for prediction:
-
Complexation free energy (kJ/mol):It is a key index to evaluate the strength of host–guest complexation and disassociation/association stability of the complex, which can decide the usage of a specific drug–CDs inclusion complex in the formulation design. Data items were the complexation free energy or the equilibrium constant by phase solubility study (T = 25 °C, pH = 7).
Figure 1: the statistic of the dataset.
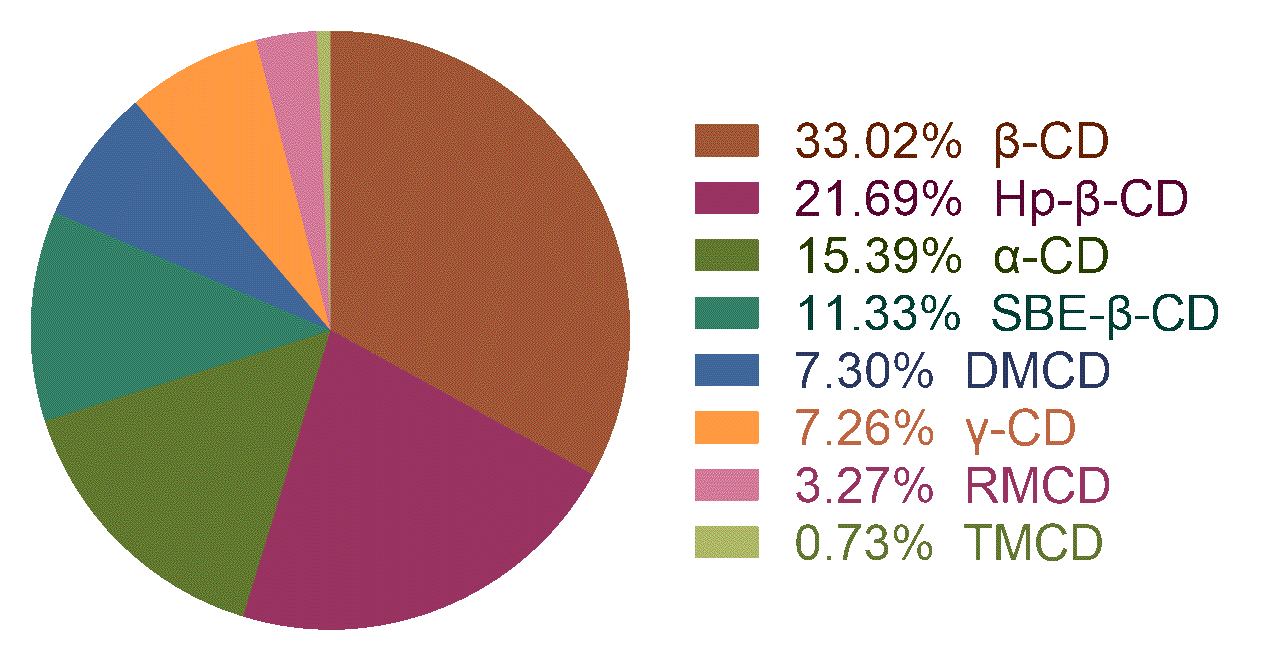
Figure 2: the performance of the model.
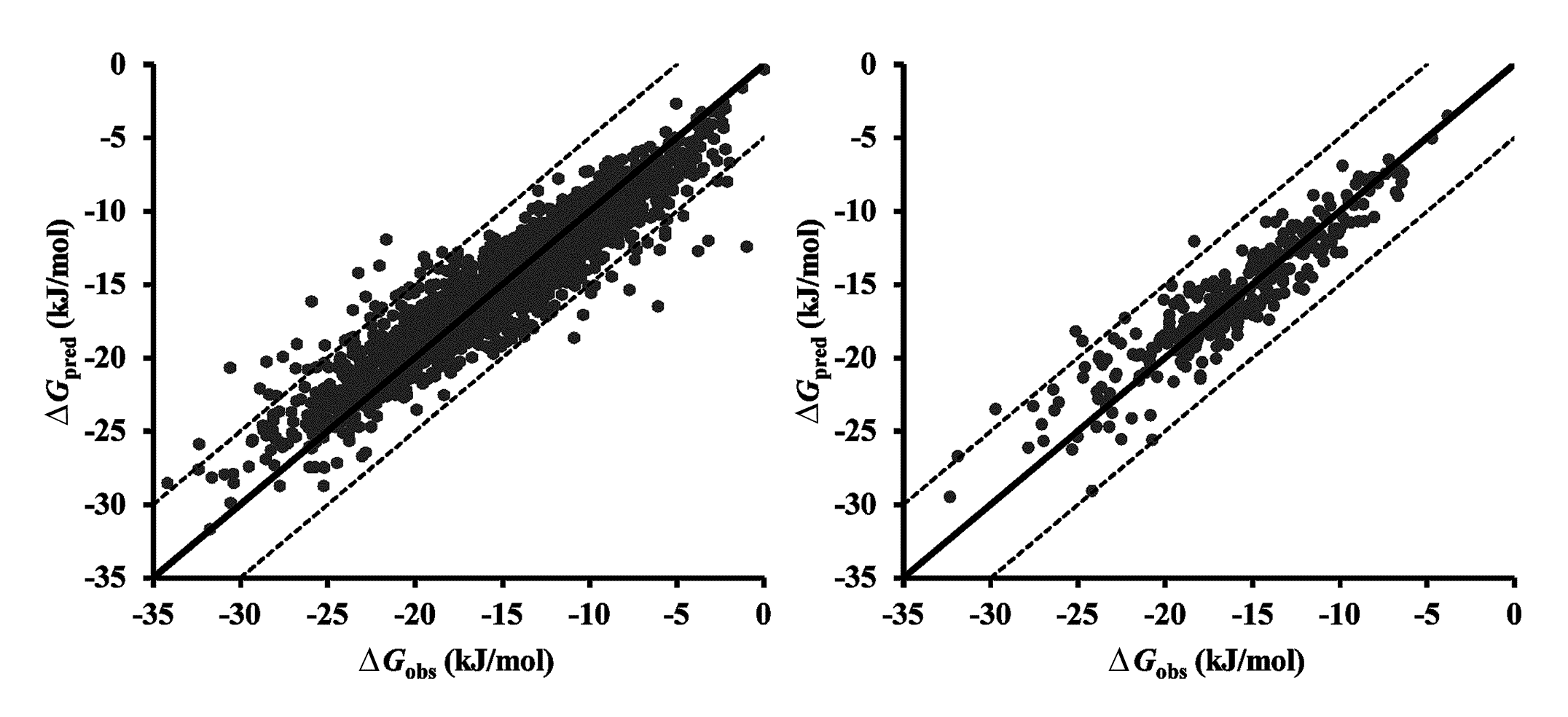
Table 1: the parameters and statistics.
| Item | Value |
|---|---|
| Training set number: | 2318 |
| Validation set number: | 339 |
| Test set number: | 335 |
| Training set R2/RMSE | 0.86/1.94 |
| Validation set R2/RMSE | 0.87/1.63 |
| Test set R2/RMSE | 0.86/1.83 |

Zhao Q, Ye Z, Su Y, et al. Predicting complexation performance between cyclodextrins and guest molecules by integrated machine learning and molecular modeling techniques[J].
Acta Pharmaceutica Sinica B, 2019, 9(6): 1241-1252.
Amorphous solid dispersion (SD)
is an effective solubilization technique for water-insoluble drugs. However,
physical stability issue of solid dispersions still heavily hindered the
development of this technique. Traditional stability experiments need
to be tested at least three to six months, which is time-consuming and unpredictable.
In this research, a novel prediction model for physical stability of solid dispersion
formulations was developed by machine learning techniques. 646 stability data points
were collected and described by over 20 molecular descriptors.
All data was classified into the training set (60%), validation set (20%),
and testing set (20%) by the improved maximum dissimilarity algorithm (MD-FIS).
Eight machine learning approaches were compared and random forest (RF) model
achieved the best prediction accuracy.
Properties for prediction:
-
Physical stability:The experimental of stability experiments with a storage time (3-month and 6-month). Data items were reported results of stable or unstable.
Figure 1: the statistic of the dataset.
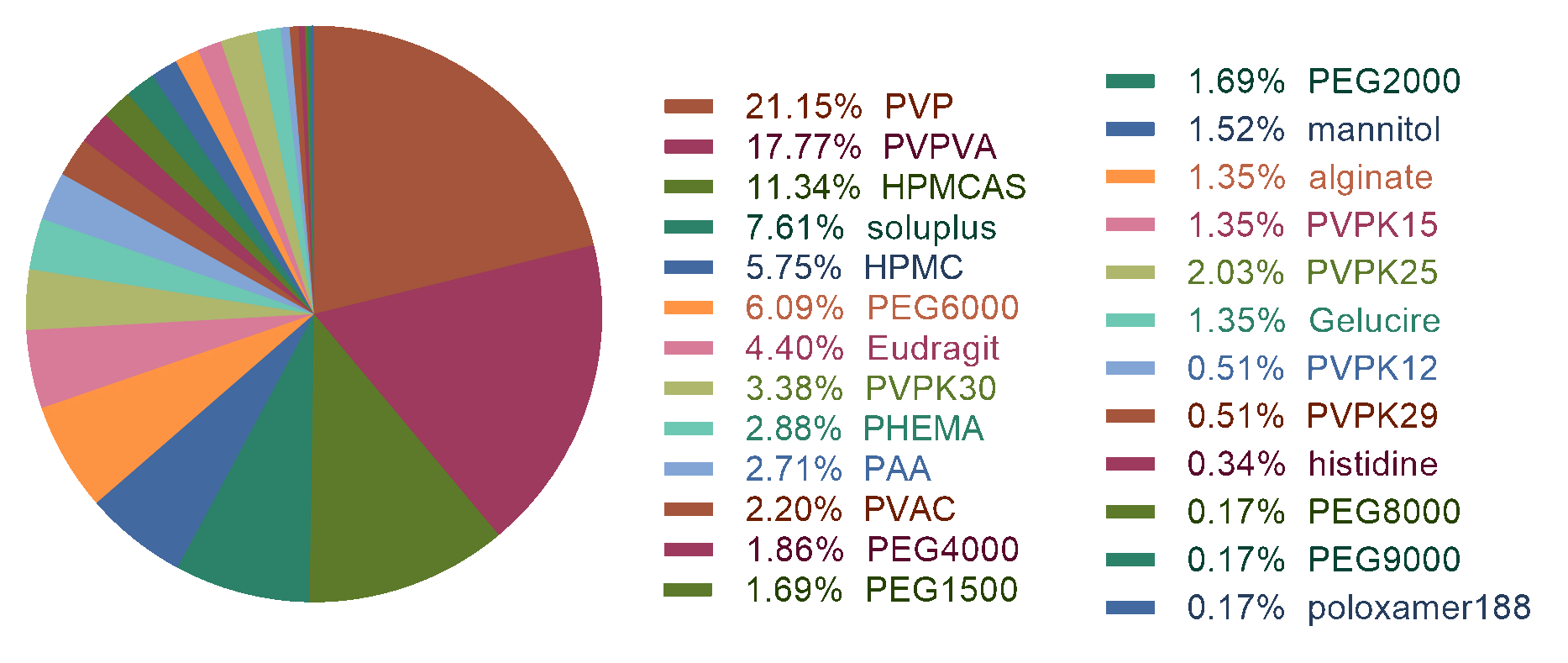
Table 1: the parameters and statistics.
| Item | 3 month | 6 month |
|---|---|---|
| Training set: | 407 | 407 |
| Validation set: | 121 | 121 |
| Test set: | 121 | 121 |
| Training set ACC | 0.951 | 0.941 |
| Validation set ACC | 0.833 | 0.817 |
| Test set ACC | 0.9 | 0.842 |

Han R, Xiong H, Ye Z, et al. Predicting physical stability of solid dispersions by machine learning techniques[J].
Journal of Controlled Release, 2019, 311: 16-25.
The drug-phospholipid complexation technique is widely used as an increasingly common method
to improve the bioavailability of drugs. This project aims to predict the complexation
rate of phospholipid complex by machine learning methods. 363 drug/phospholipid complex
formulations were collected from the literature. The datasets were trained by several popular
machine learning methods including RandomForest, SVM, KNN, Naive Bayes, Decision Tree, XGBoost, LightGBM.
By analysis and compare these methods, the RandomForest got better performance.
The predictive accuracy and AUC of RandomForest
was 0.772/0.824 on training set(5-fold cross validation), 0.808/0.902 for the test set, respectively.
Properties for prediction:
-
Phospholipid complexation:Phospholipids and candidate drugs can form complexation with hydrogen bonds or the van der Waals interaction in the alcoholic or organic solvent under certain conditions. The experimental data from literature were considered acceptable when they were binary phospholipid complex with complexation rate or other characterization.The complexation was considered successful only if the complexation rate was above or equal 80%. If less than 80%, it was considered unsuccessful.
Figure 1: the statistic of the phospholipids in dataset.
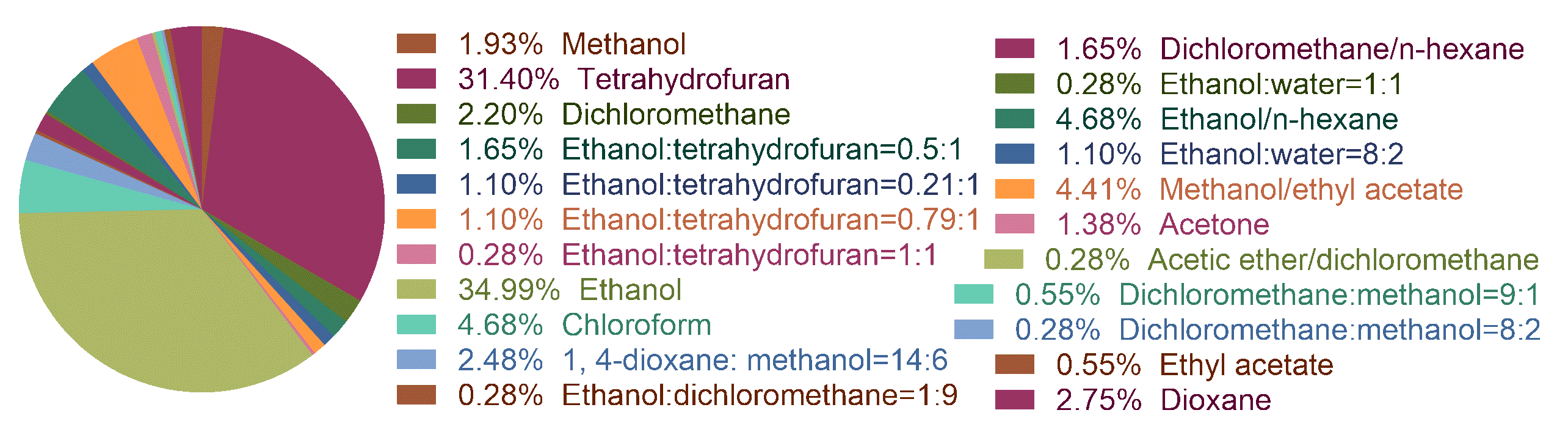
Table 1: the parameters and statistics.
| Item | Combination |
|---|---|
| Training set: | 290 |
| Test set: | 73 |
| Training set ACC/AUC | 0.772/0.824 |
| Test set ACC/AUC | 0.808/0.902 |

Gao H, Ye Z, Dong J, et al. Predicting drug/phospholipid complexation by the lightGBM method[J].
Chemical Physics Letters, 2020, 747: 137354.
Nanocrystals have exhibited great advantages for solving the dissolution issue of water insoluble drugs.
In this study, the machine learning models for the three nanocrystal preparation methods were
constructed for the nanocrystal size and PDI predictions. The results demonstrated
that random forest (RF) exhibited well predictive performance for the ball wet milling (BWM),
high-pressure homogenization (HPH) methods and anti solvent method (ASP).
Here, by submitting a molecule, we can predict both size and PDI for the three methods.
Properties for prediction:
-
Size (nm):Particle size of drug nanocrystals. -
PDI:Polydispersity of drug nanocrystals index. It is a parameter used to reflect the degree of non-uniformity of size distribution of particles. The numerical value of PDI ranges from 0 to 1.
Figure 1: the statistic of the antisolvent dataset.
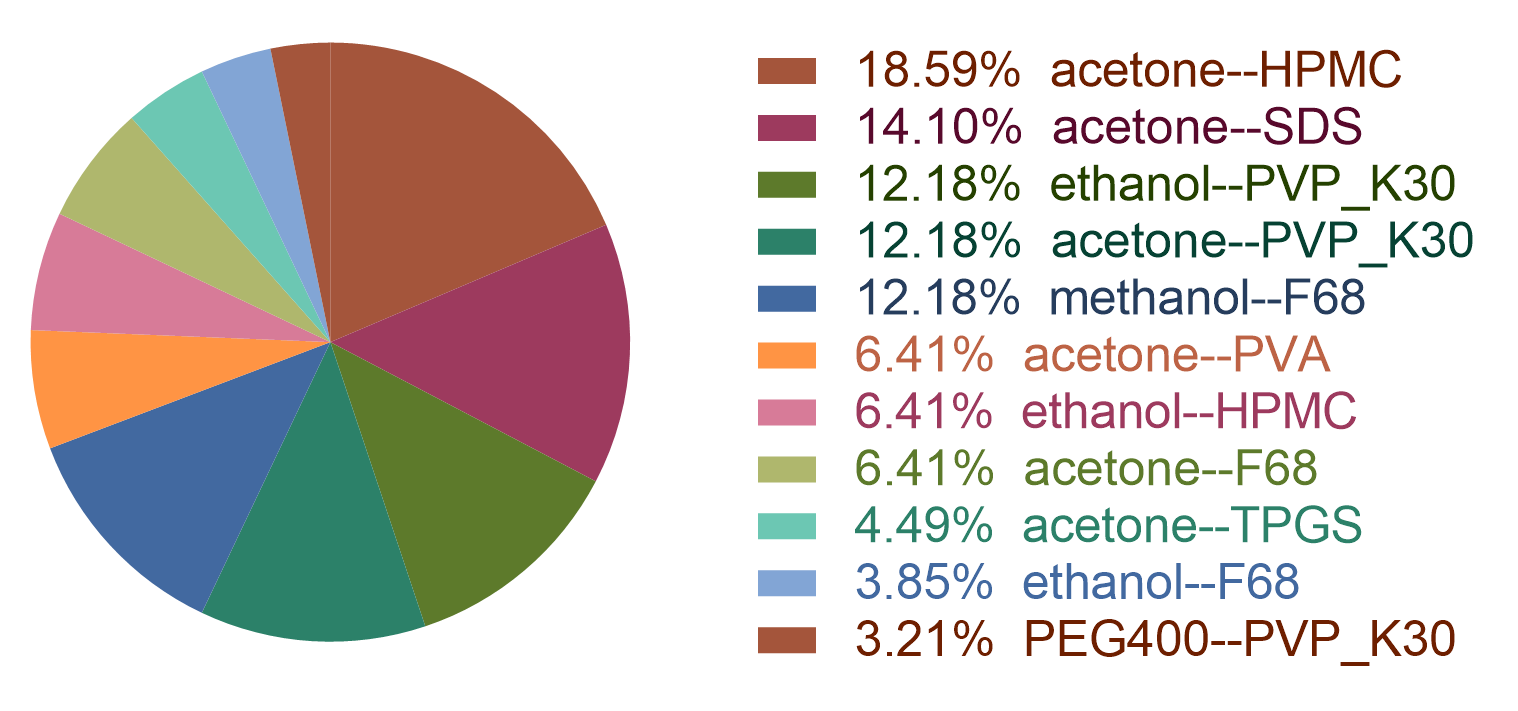
Figure 2: the statistic of the HPH dataset.
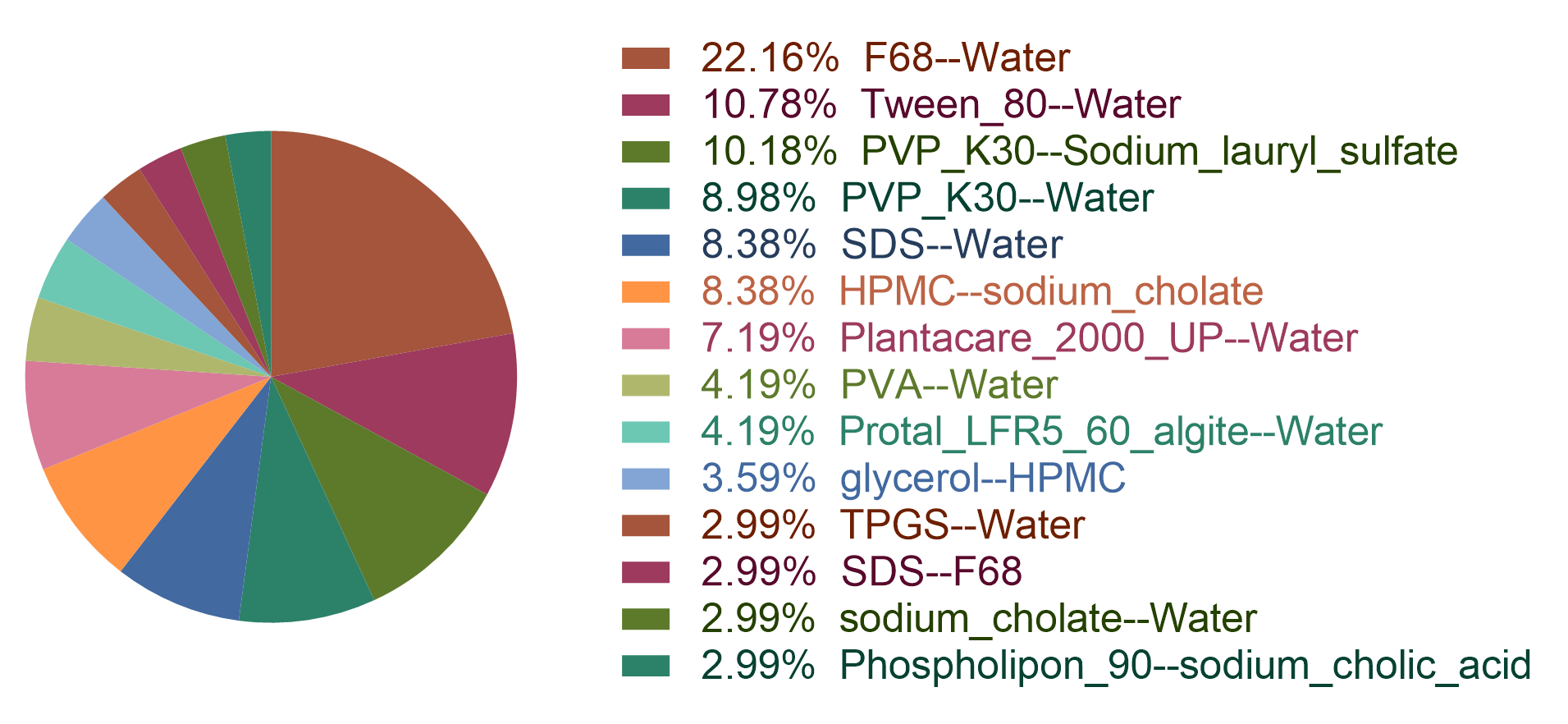
Figure 3: the statistic of the BWM dataset.
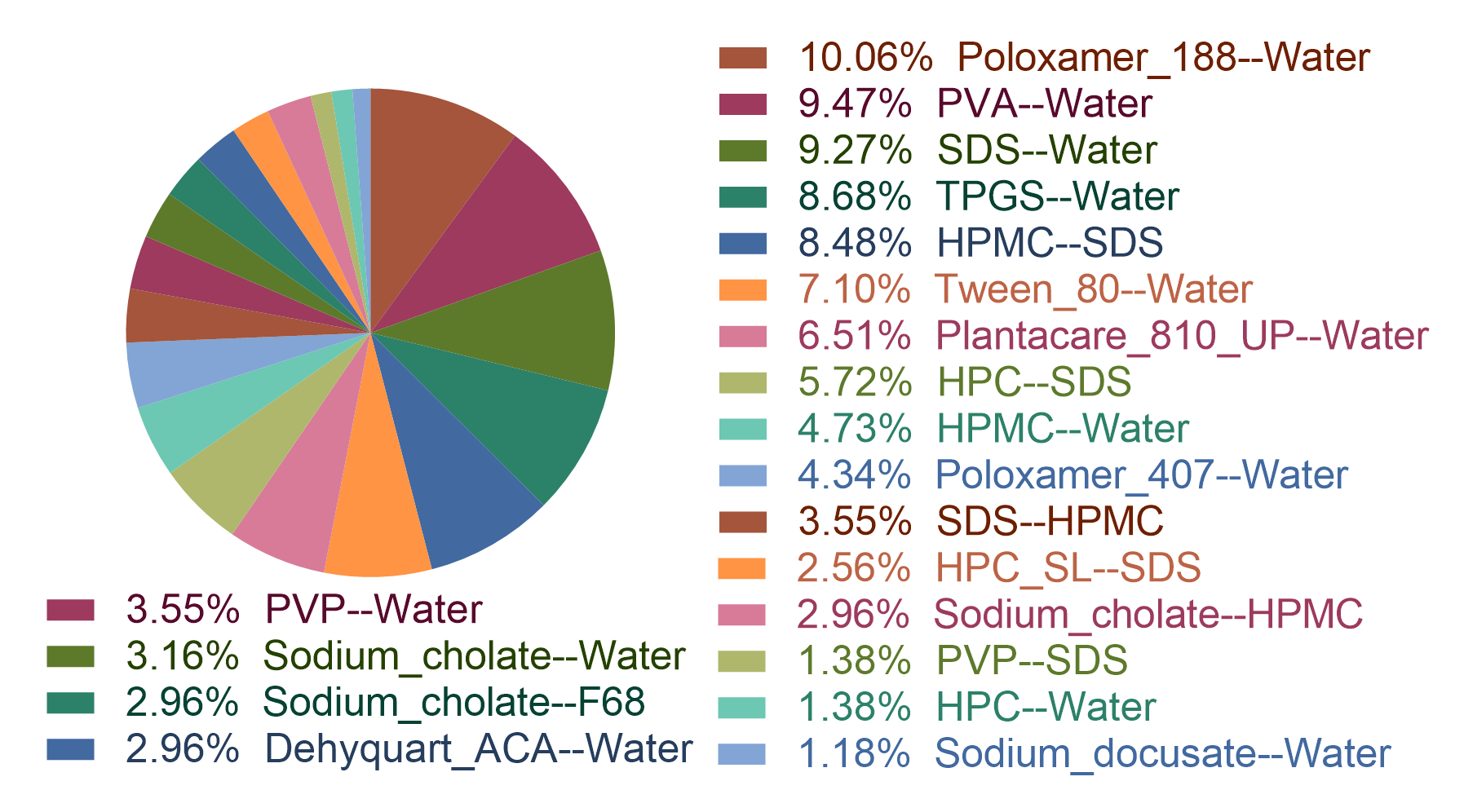
Table 1: the statistics of the models.
| Dataset | Size(Qcv2/Rt2) | PDI(Qcv2/Rt2) |
|---|---|---|
| Antisolvent: | 0.434/0.419 | 0.593/0.518 |
| HPH: | 0.585/0.554 | 0.786/0.690 |
| BWM: | 0.699/0.786 | 0.837/0.796 |
He Y, Ye Z, Liu X, et al. Can machine learning predict drug nanocrystals?[J].
Journal of Controlled Release, 2020, 322: 274-285.
Self-emulsifying drug delivery system (SEDDS), a thermodynamic stable nano formulation, is consists of
oil,
surfactant, co-surfactant and APIs. For oral administration,
drugs are dissolved in the SEDDS instead of solid state,
which benefit to the absorption in the Gi tract.
Here, a pseudo-ternary phase diagram predicting model was constructed by random forest algorithm
and this model could predict the self-emulsion status of the formulation under specified conditions.
Properties for prediction:
-
SEDDS status:It is an endpoint represents the self-emulsifying status of each point in pseudo-ternary phase diagram. The process of SEDDS formulation design includes three step as below: the determination of drug solubility in several oils, surfactants and cosurfactants; dissolve the mixture of oils, surfactants and cosurfactant into distilling water, and then draw the ternary phase diagram to identify the self-emulsion area.In the self-emulsion area, the coordinate point inside the intersection point between the self-emulsifying region and the grid lines were selected as the data points of self-emulsion. In the non-self-emulsifying area, the vertex of the grid line was selected as the data point of non-self-emulsion. Each selected point in the pseudo-ternary phase diagram was a single data point.
Figure 1: the statistic of the oli types in the dataset.
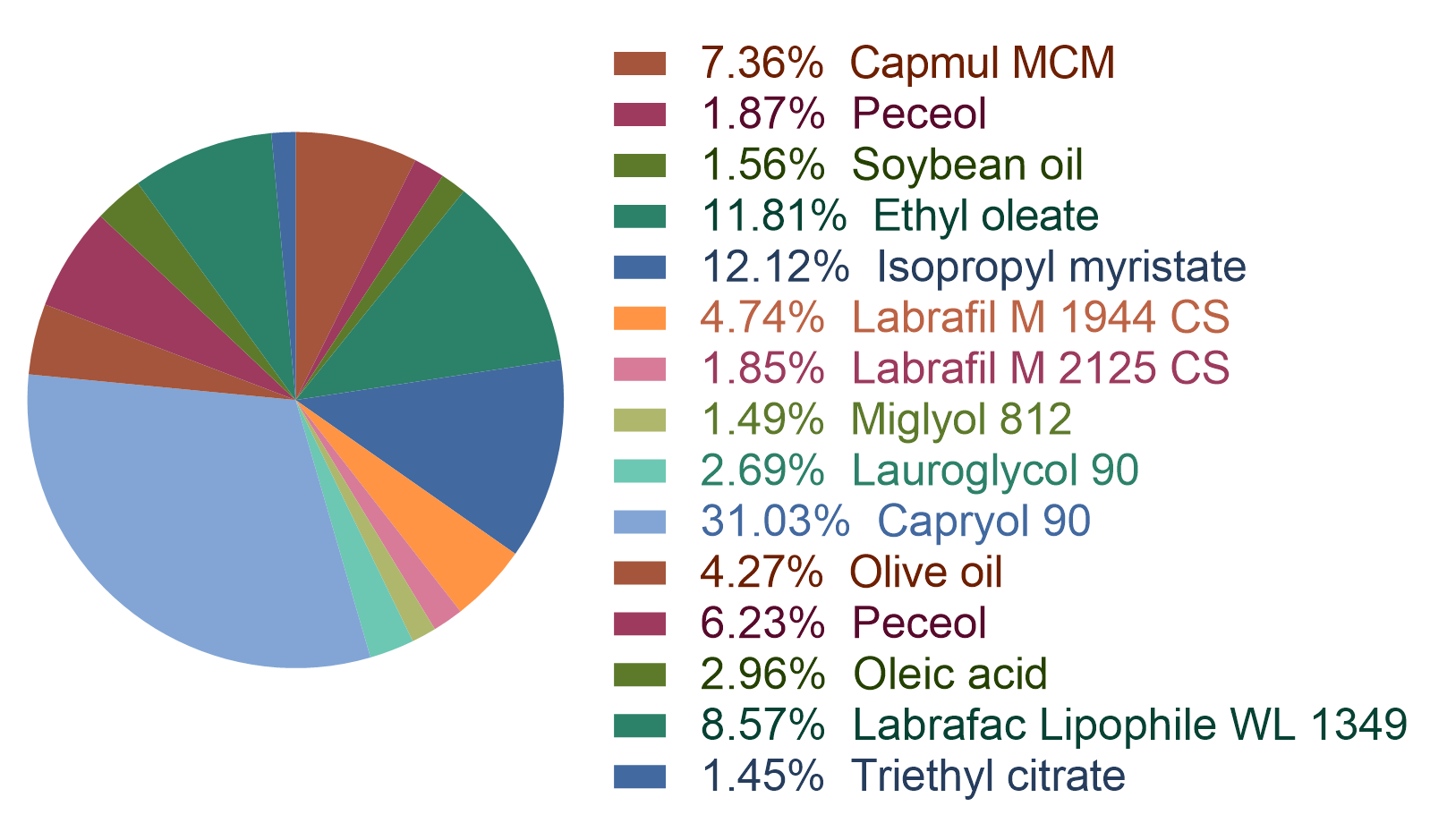
Figure 2: the statistic of the surfactant types in the dataset.
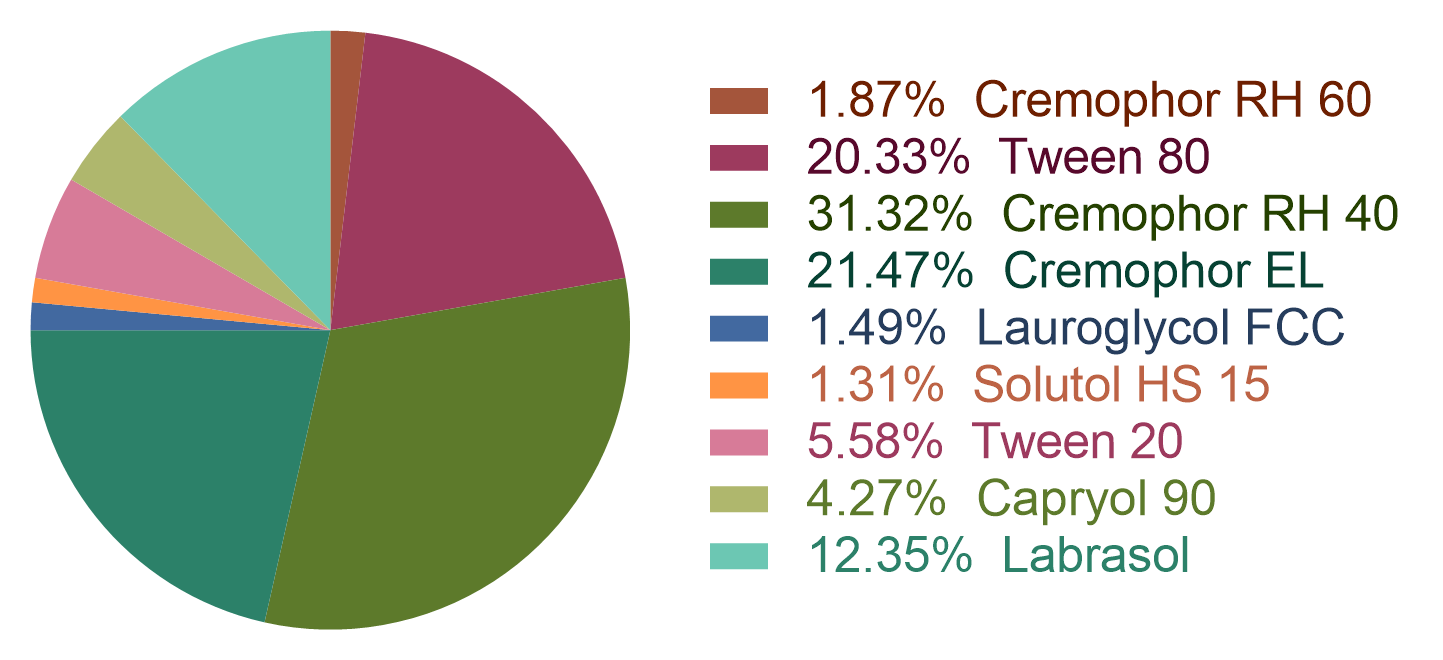
Figure 3: the statistic of the cosurfactant types in the dataset.
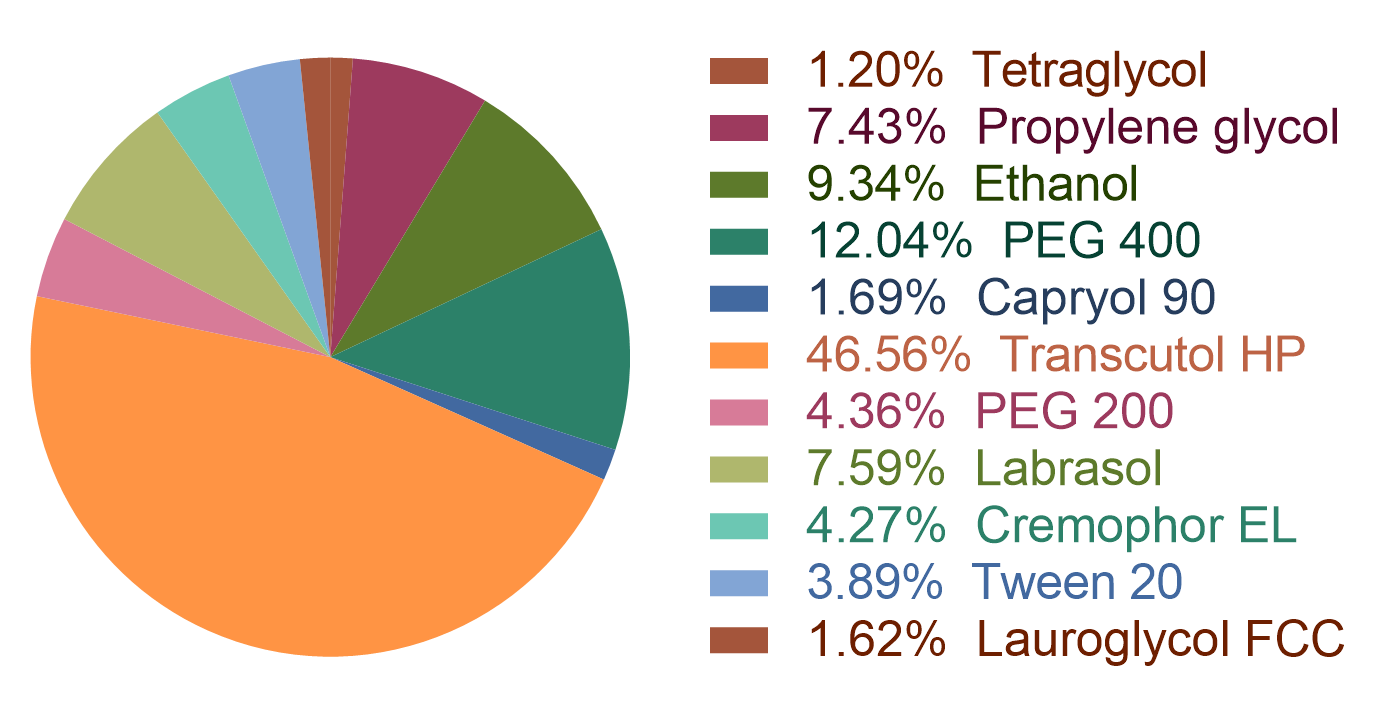
Table 1: the statistics of the models.
| ACC(5-fold CV/Test) | AUC(5-fold CV/Test) | SE(5-fold CV/Test) | SP(5-fold CV/Test) |
|---|---|---|---|
| 0.925/0.926 | 0.977/0.978 | 0.922/0.927 | 0.927/0.926 |
Gao H, Jia H, Dong J, et al. Integrated in silico formulation design of self-emulsifying drug delivery systems[J].
Acta Pharmaceutica Sinica B, 2021, 11(11): 3585-3594 (JCRC Q1, IF: 14.90).
Liposome is a spherical vesicle that consists of a bilayer of lipid and internal
hydrophilic cavity with particle sizes ranging from 30 nm to several micrometers.
As its great biocompatibility, biodegradability and low toxicity, liposome
is regarded as a promising drug delivery system, not only for drug molecule delivery,
but for genes and bio-macromolecules. The composition of lipids, particle size,
surface charge and lamellarity would decide the structure and morphology of liposome
particles. The various preparation methods have been applied in liposome products.
With the development of liposome technology, a number of liposome-based drug formulations
have been available for market and for clinic trails. In current research, size, PDI,
zeta potential and encapsulation of liposome are regarded as key parameters for formulation
evaluation. Herein, we develop a prediction platform for
these key parameters of liposome.
Properties for prediction:
-
Size (nm):The size of the liposome formulation. -
PDI:The polydispersity index of the liposome. -
Zeta (mV):zeta potential is the electric potential in the interfacial double layer of a dispersed particle or droplet versus a point in the continuous phase away from the interface. -
Encap (%):It represents the efficiency or the percentage of drug that is successfully entrapped into the formulation.
Table 1: the statistics of the models.
| Properties | Qcv2/Rt2 | RMSEcv/RMSEt | MAEcv/MAEt |
|---|---|---|---|
| Zeta: | 0.571/0.644 | 17.528/12.559 | 11.086/8.299 |
| PDI: | 0.408/0.604 | 0.081/0.055 | 0.059/0.042 |
| Size: | 0.707/0.823 | 145.271/116.002 | 75.263/67.198 |
| Encap: | 0.764/0.724 | 16.185/17.926 | 10.853/11.333 |
Drug solubility is a basic property for the formulation design, especially in different
solvents. Here, we collected a current largest dataset containing more than 4000 data points.
Then, we employed lightGBM and RandomForest algorithms to build a robust model. This tool enables users
to predict drug solubility in 27 kinds of solvent.
Properties for prediction:
-
Drug solubility (mol/L):It is an endpoint represents the solubility of durg molecules in different solvents (T = 25 °C).
Table 1: the performance of the model.
| Items | Values |
|---|---|
| Qcv2 | 0.922 |
| RMSEcv | 0.380 |
| MAEcv | 0.180 |
| Rt2 | 0.910 |
| RMSEt | 0.395 |
| MAEt | 0.177 |
Figure 1: the statistics of the solvents.
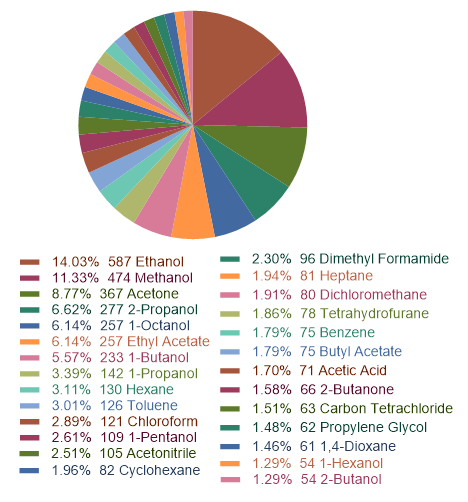

Ye Z, Defang Ouyang. Prediction of small-molecule compound solubility in organic solvents by machine learning algorithms[J].
Journal of Cheminformatics, 98(2021).
Copyright @ 2018-2019 Computational Pharmaceutical Group,
State Key Laboratory Of Quality Research in Chinese Medicine, ICMS, University of Macau.
All rights reserved.
The recommended browsers: Safari, Firefox, Chrome,IE(Ver.>8).
FormulationAI is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.  E-mail: jiedong@csu.edu.cn
E-mail: jiedong@csu.edu.cn
 |
 |
 |
 |
|
 |